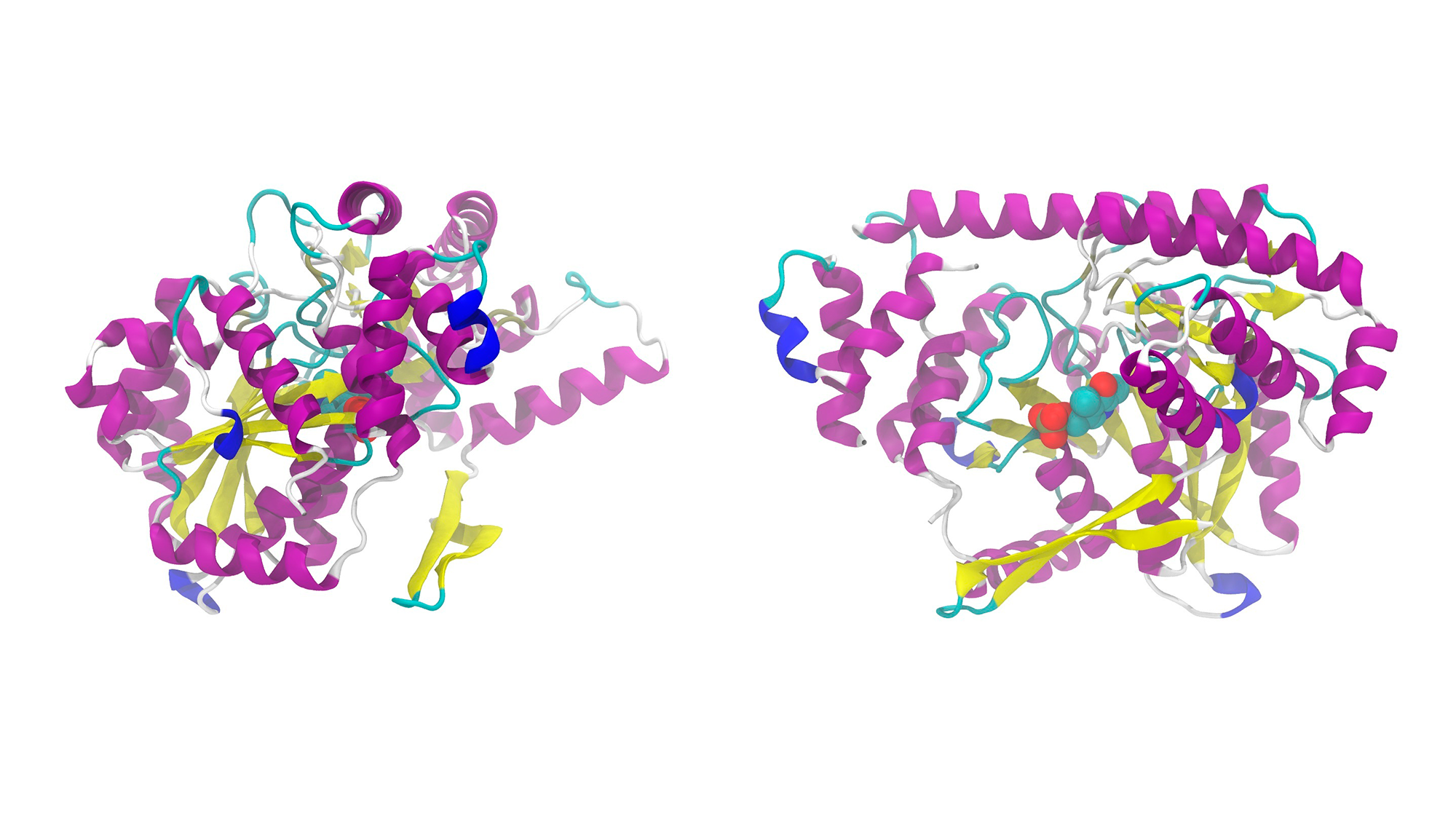
A study carried out by the Centre for Genomic Regulation (CRG) and the Shenzen-based company BGI has determined that missense mutations in DNA that cause diseases do so mostly because they destabilise the protein they encode. The team observed this in around 60% of the mutations they studied – by introducing them into yeast cells – and it is because the mutation results in misfolding of the encoded protein.
However, the team led by Ben Lehner also observed several mutations that do not destabilise the proteins, but also cause diseases. This opens the door to differential treatments depending on the consequences of the mutation. Some of diseases they studied were heritable cataracts, as well as developmental, muscle-wasting or neurological diseases, such as Rett Syndrome.
This study was made possible thanks to the Human Domainome 1 project, which has catalogued more than half a million mutations in around 500 human protein domains. These mutations were created by systemically changing every amino acid in these domains for every other possible amino acid. Although it is the largest such catalogue to date, it covers only 2.5% of the known human proteins.
From diseases to the tree of life
In other research from CRG, led by Cedric Notredame, protein structure data have been combined with genomic sequence data to better determine phylogenetic trees. These are key to understanding the evolution of life on earth, as well as to monitoring the evolution of pathogens o developing new treatments.
Traditionally, phylogenetic trees have been created only with DNA or protein sequence data and looking at the similarities and the differences between ancestors. Unfortunately, changes between sequences over time can be so great that the line between ancestral and modern forms get lost.
Nevertheless, despite mutations, protein structure remains fairly constant. Here, the study of computationally predicted structure changes, especially the distance between amino acids, has been combined with sequence changes to draw more precise evolutionary trees. Artificial intelligence has also been needed to predict the protein structures, as well as providing hitherto unknown data.
Beltran, A., Jiang, X., Shen, Y. et al. Site-saturation mutagenesis of 500 human protein domains. Nature 637, 885–894 (2025). https://doi.org/10.1038/s41586-024-08370-4
Baltzis, A., Santus, L., Langer, B.E. et al. multistrap: boosting phylogenetic analyses with structural information. Nat Commun 16, 293 (2025). https://doi.org/10.1038/s41467-024-55264-0