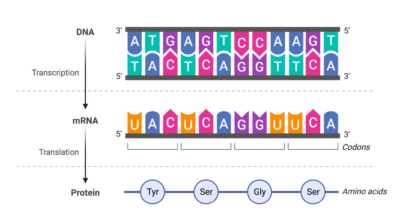
Until recently, it was assumed that one mutation could often affect another mutation, increasing or suppressing its effects. Now, a study by the Centre for Genomic Regulation (CRG) in collaboration with the Wellcome Sanger Institute has found that interactions between mutations are less common than previously thought. This means that most mutations affect a protein independently.
The discovery that mutations usually do not interact with each other implies that protein stability is affected by mutations following much simpler rules than previously assumed. The research team, led by André Faure and Ben Lehner, analyzed protein sequences with different combinations of mutations and tested their stability. The experimental result closely matched models that calculated the overall effect of mutations as a simple sum, considering them independent. However, the model was able to predict the protein’s structure if it had less than three mutations.
“We’ve seen that we don’t need supercomputers to predict a protein’s behavior; simple measurements and maths will suffice.”
Ben Lehner (CRG and Wellcome Sanger Institute)
Nevertheless, some level of experimental validation is still required to confirm predictions, especially for critical applications such as drug development, where unexpected effects or rare interactions may not be captured by the models. But this research will help optimize the number of experiments needed to analyze and predict protein structures, which represents a step forward in designing proteins with pharmaceutical and biotechnological potential.
Faure, A.J., Martí-Aranda, A., Hidalgo-Carcedo, C. et al. The genetic architecture of protein stability. Nature (2024). https://doi.org/10.1038/s41586-024-07966-0