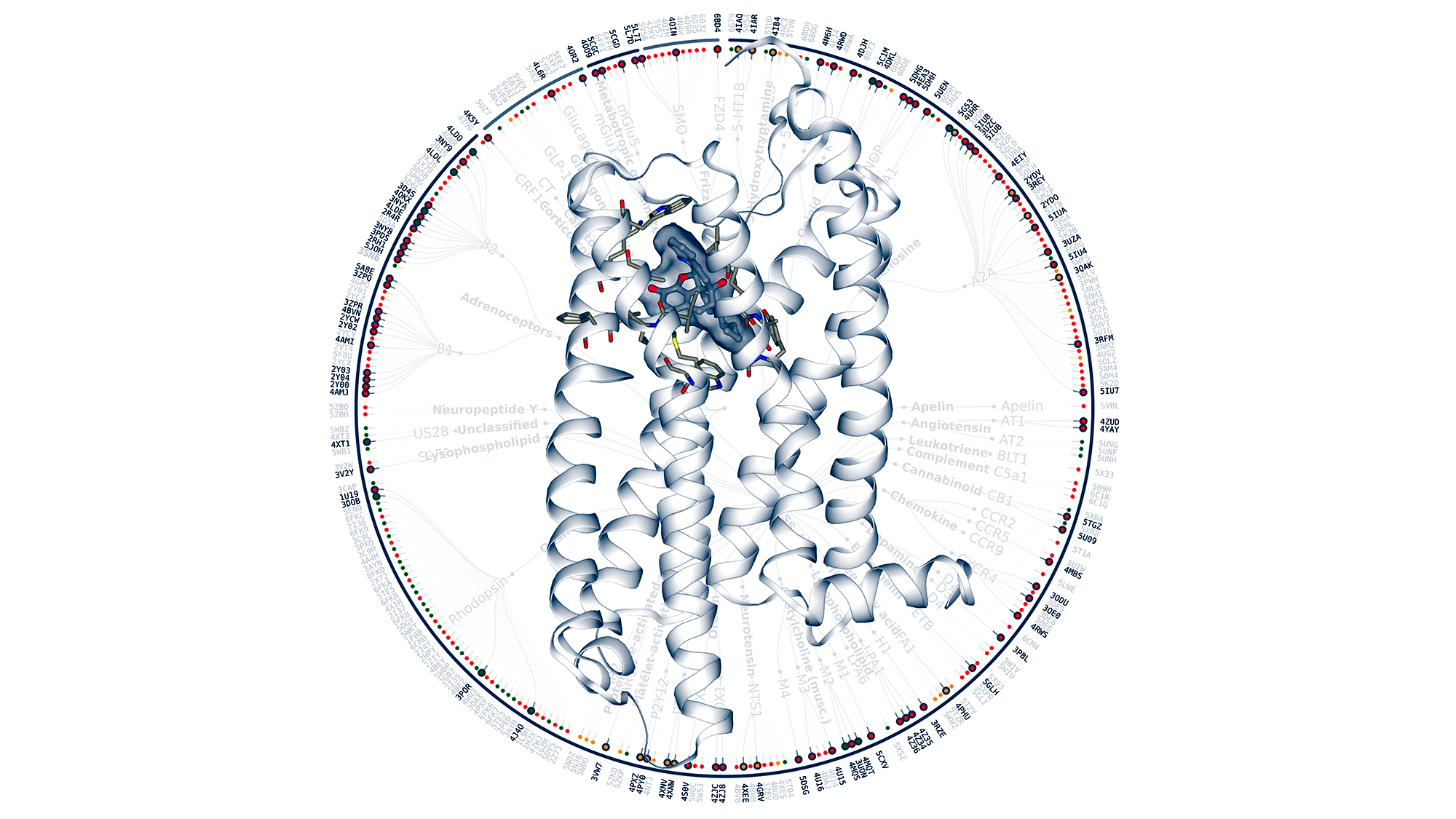
In our body there are more than 800 G protein-coupled receptors (GPCRs), proteins which present in the cell membrane and that are responsible for transmitting signals within the cell. They are involved in many processes – and in diseases from Alzheimer’s or Parkinson’s to cancer. That is why they are targets for almost 40% of current drugs.
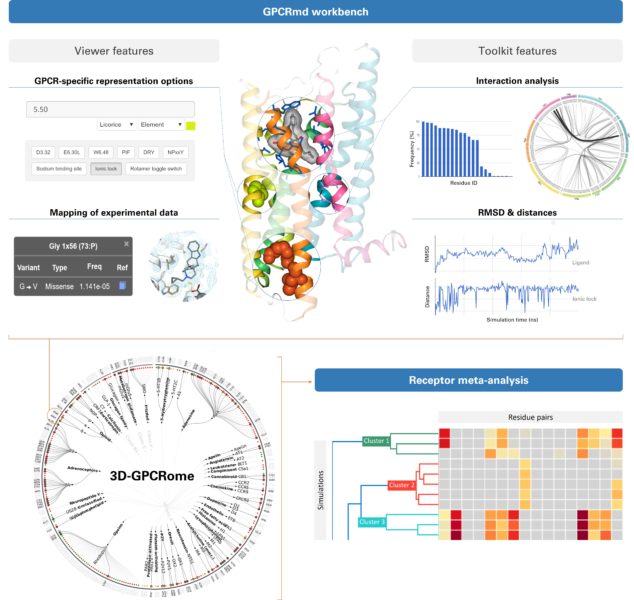
A consortium of 24 laboratories from 10 countries has just released a platform called GPCRmd for scientists from different disciplines so they can easily inspect and analyze a comprehensive number of molecular simulations of GPCRs. This could assist in the design of new drugs that have GPCRs as targets or in personalized medicine.
The initiative has been led by the GRIB, joint research programme of the Hospital del Mar Medical Research Institute (IMIM) and the Department of Experimental and Health Sciences of the Universitat Pompeu Fabra, with significant support from the Paul Scherrer Institute in Switzerland,
The GPCRmd platform allows an exhaustive number of molecular simulations of GPCRs to be easily analyzed. This could assist in the design of new drugs or in personalized medicine.
We speak with Mariona Torrens Fontanals, one of the main authors of this work. The young scientist is a PhD student in the GPCRs-based drug development research group of the Biomedical Informatics Research Program (GRIB), led by Jana Selent, a Miguel Servet researcher at IMIM. Both are in charge of managing the GPCRmd platform.
Interview to Mariona Torrens Fontanals

What are GPCRs and why are they so important?
They are proteins that are found in the membrane of many cells in our body and are responsible for transmitting a signal to the interior of the cell. There are many different types – it is the largest family of proteins in humans! So they are involved in many different processes. In fact, 40% of the drugs on the market have a GPCR as a target!
What difficulties do they entail when developing drugs against them?
They are very complex proteins. They can cause more than one signal at the same time – one may be the therapeutic effect you are looking for, but the other may be a side effect you don’t want. And it is difficult to know how to activate the protein so that it does only what you want. Their structure varies according to which molecule binds to them. And when their structure varies, the signaling they send inside the cell varies. Thus, depending on how a potential drug binds to it, it can have one action or another. This is why understanding its structure is so important, and in the last 10 years the 3D structure of many GPCRs has been deciphered.
But knowing its static structure is not enough…
It is one thing to determine the structure by crystallography or other techniques. But in the case of GPCRs, the structure is very dynamic, and so what is interesting is to then make simulations with molecular dynamics (DM) to explore the entire conformational landscape of each GPCR at the atomic level – that is, to look at atom by atom and see what forces are applying to each other at every moment. We do this every 2 femtoseconds (0.000 000 000 000 000 002 seconds) and we see how the protein moves, how its structure changes, atom by atom. Now, this requires a high level of experience and technical knowledge as well as a lot of computer space and specialized software…
Molecular dynamics (DM) simulations allow the entire conformational landscape of each GPCR to be explored atom by atom, looking at what forces are being applied to each other every 2 femtoseconds.
And out of this need GPCRmd was born.
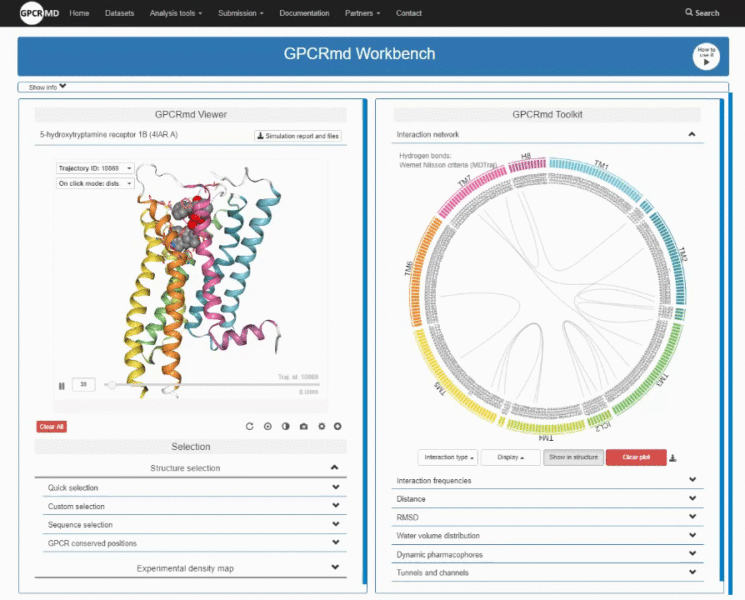
Yes, as a tool to share all these simulations with the scientific community. Not only between bioinformaticians or structural biologists – who are the ones who do molecular dynamics – but with researchers from different disciplines interested in GCPRs at the experimental level: people who are developing drugs, who want to understand how a certain mutation or variant in a GPCR modifies its action… For all these people, the information provided by molecular dynamics simulations is very useful, but often they do not use it because they do not know where to get it or how to interpret it. GPCRmd is an open platform where you can find these simulations of GPCRs. At the moment there are 70% of all the GPCRs for which the structure is known, but we hope to reach 100%. And in addition to these simulations, there are also analysis examples that can be done so that anyone interested can get the most out of this data.
Here you can listen to Mariona Torrens Fontanals telling in first person how and why they have created GPCRmd (video in Catalan)
Rodriguez-Espigares et al. GPCRmd unveils the dynamics of the 3D-GPCRome. 2020, Nature Methods; https://www.nature.com/articles/s41592-020-0884-y